![]()
StaVia - Multi-Omic Single-Cell Cartography for Spatial and Temporal Atlases
StaVia (Via 2.0) is our new single-cell trajectory inference method (new paper released) that explores single-cell atlas-scale data and temporal and spatial studies enabled by:
Graph augmentation integrating metadata (spatial omics and time-series studies): Using sequential metadata (temporal labels, hierarchical information, spatial coordinates) to guide the cartography
Higher Order Random Walks: Leveraging higher order random walks with memory of a cell’s past states to highlight key end-to-end differentiation pathways along the atlas
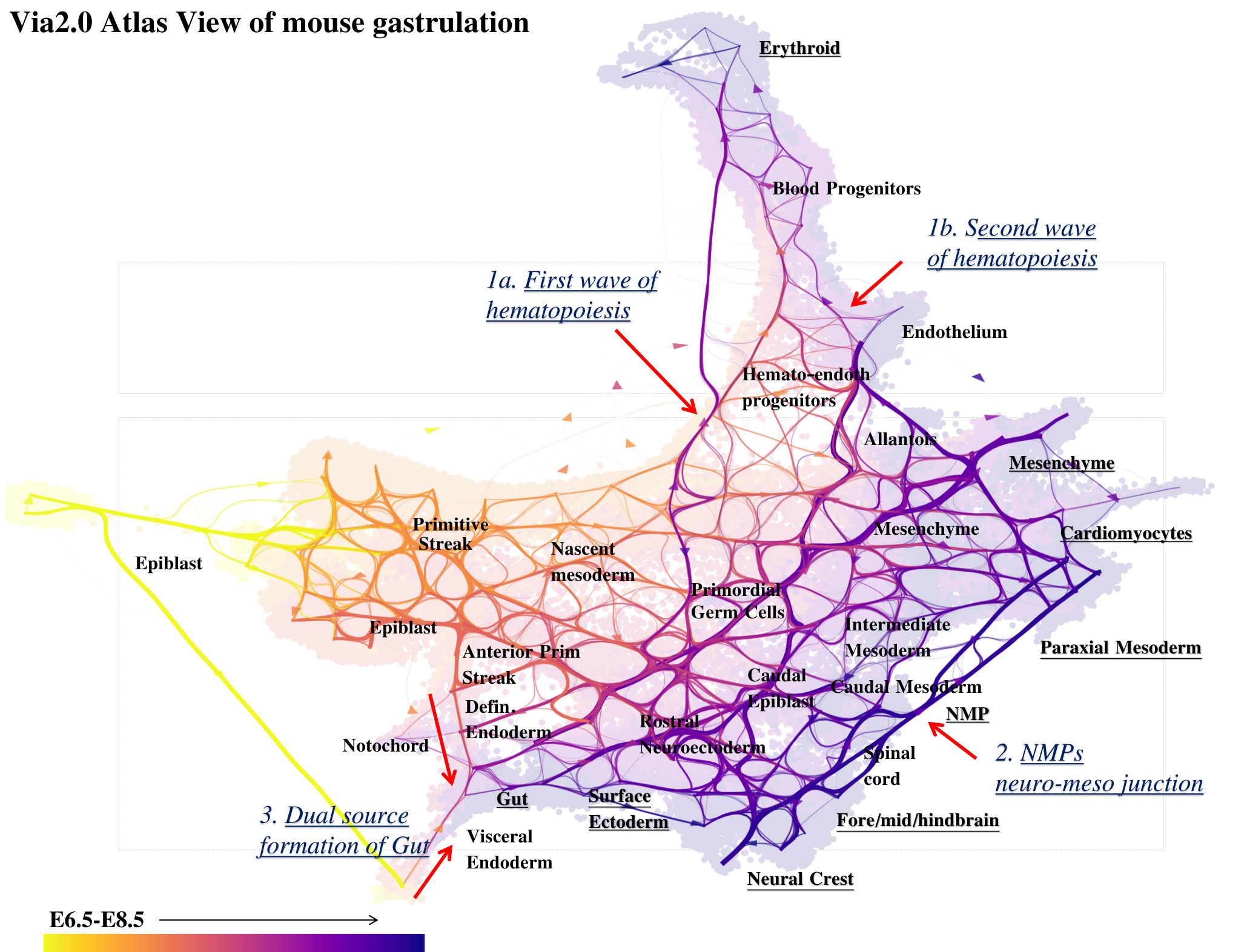
Atlas View: Via 2.0 offers a unique visualization of the predicted trajectory by intuitively merging the cell-cell graph connectivity with the high-resolution of single-cell embeddings.


StaVia still offers all the functionality of Via 1.0 . in terms of various types of topology construction (disconnected, cyclic), pseudotimes, automated terminal state prediction and automated plotting of temporal gene dynamics along lineages, for details please refer to our preprint.
StaVia extends the lazy-teleporting walks to higher order random walks with memory to allow better lineage detection, pathway recovery and preservation of global features in terms of computation and visualization. StaVia is generalizable to multi-omic analysis: in addition to transcriptomic data (time-series studies, spatial omics), VIA works on scATAC-seq, flow and imaging cytometry data.
Try out the following with StaVia:
Combining temporal information with scRNA velocity temporal study
Constructing the Atlas View visualization . H5ad anndata objects are provided on the github page
Using spatial information of cells’ location on tissues to guide the TI. See tutorials for MERFISH and Stereoseq data.
Datasets used in tutorials can be found on the github-data page
StaVia Cartographic Atlas View for Mouse Gastrulation using time-series and RNA velocity adjusted graphs